Publicado in Henry Rzepa's Blog
Autor Henry Rzepa
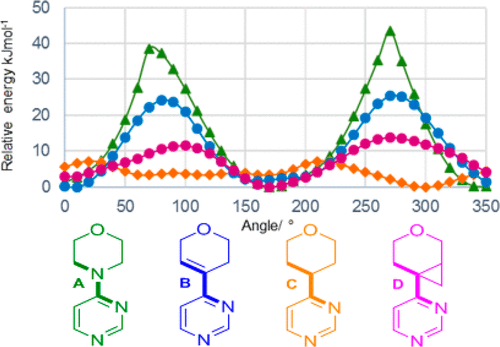
In the pipeline reports on an intriguing new ring system acting as an isostere for morpholine. I was interested in how the conformation of this ring system might be rationalised electronically and so I delved into the article.[cite]10.1021/acs.jmedchem.9b00348[/cite] Here I recount what I found.