Publicado in Henry Rzepa's Blog
Autor Henry Rzepa
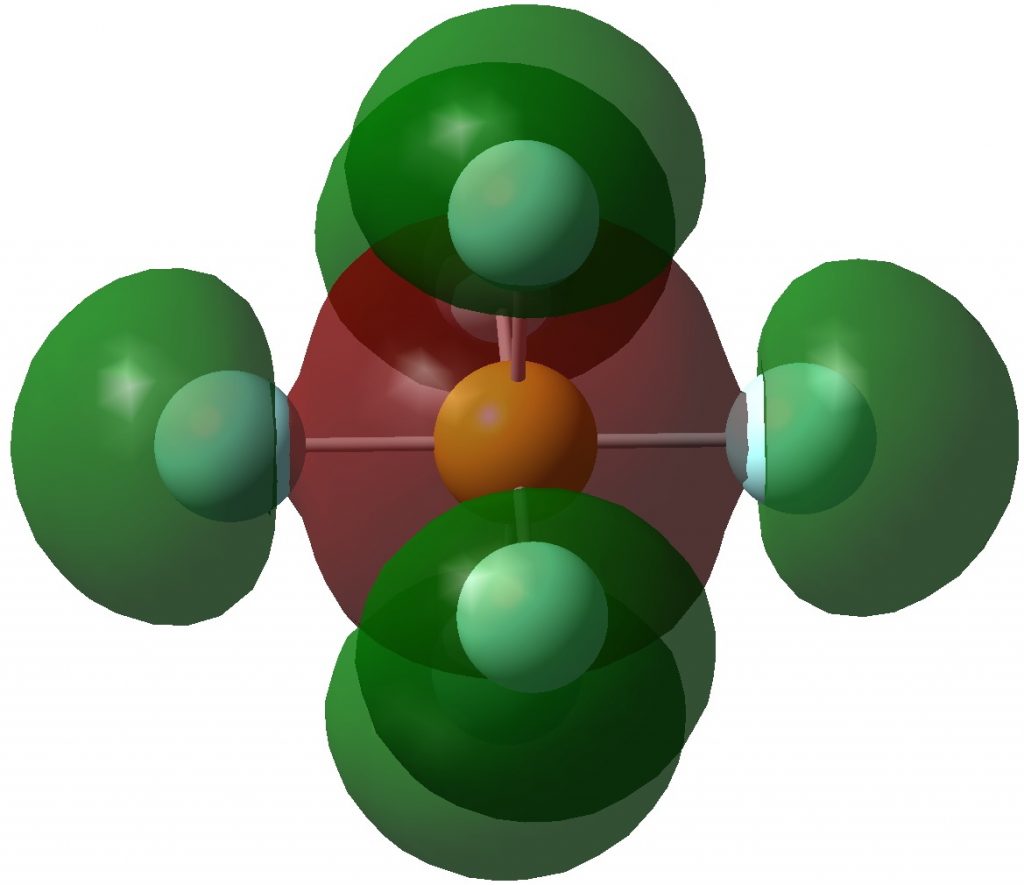
Following on from discussing octet expansion in species such as SeMe 6 , ClMe 3 and ClMe 5 , I felt impelled to return to SF 6 , often used as an icon for hypervalence.
Following on from discussing octet expansion in species such as SeMe 6 , ClMe 3 and ClMe 5 , I felt impelled to return to SF 6 , often used as an icon for hypervalence.
A few years back, I took a look at the valence-shell electron pair repulsion approach to the geometry of chlorine trifluoride, ClF 3 using so-called ELF basins to locate centroids for both the covalent F-Cl bond electrons and the chlorine lone-pair electrons.

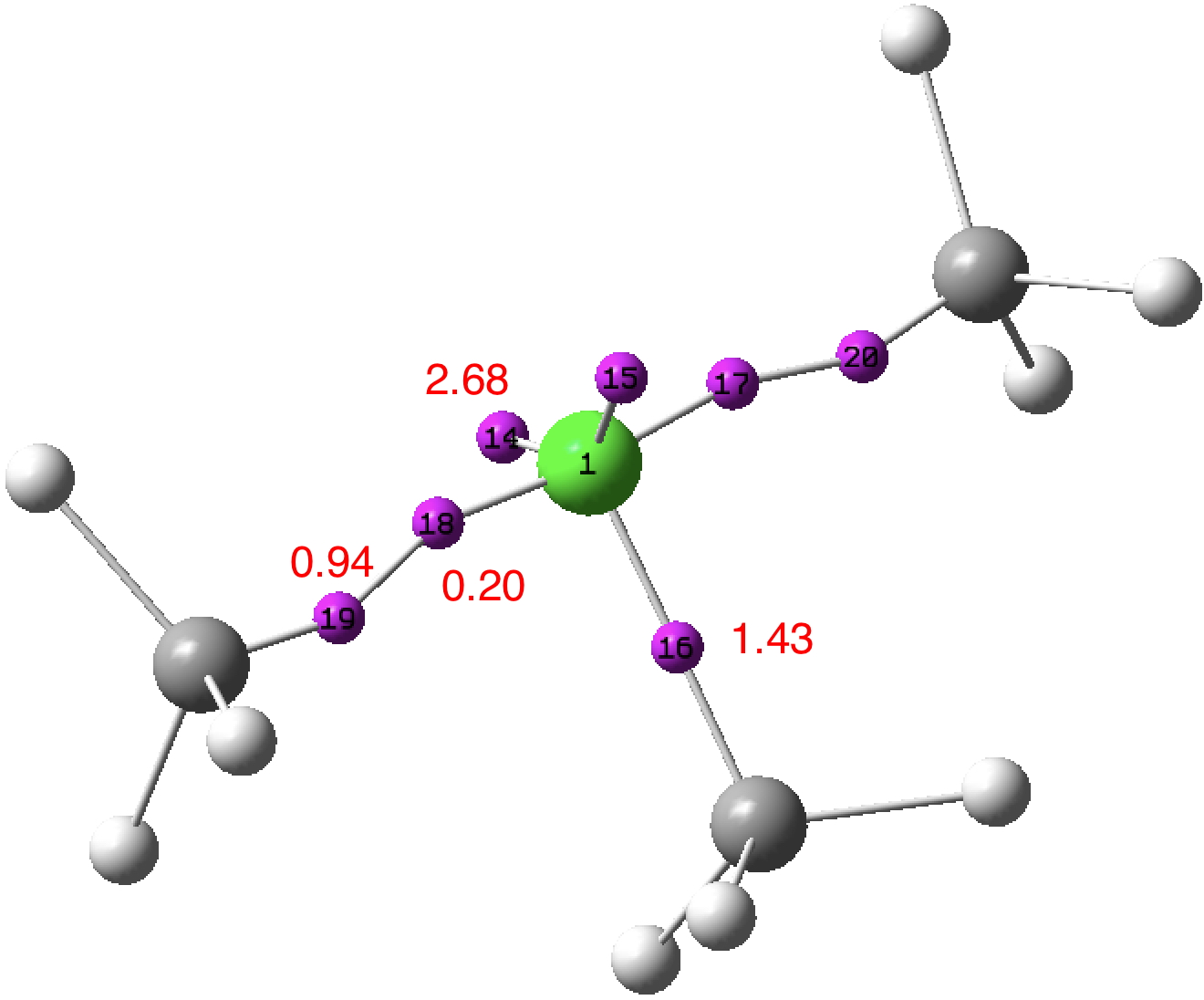
A recent article reports, amongst other topics, a computationally modelled reaction involving the capture of molecular hydrogen using a substituted borane (X=N, Y=C).[cite]10.1073/pnas.1709586114[/cite] The mechanism involves an initial equilibrium between React and Int1 , followed by capture of the hydrogen by Int1 to form a 5-coordinate borane intermediate ( Int2 below, as per

Early in 2011, I wrote about how the diatomic molecule Be 2 might be persuaded to improve upon its normal unbound state (bond order ~zero) by a double electronic excitation to a strongly bound species.
Another selection (based on my interests, I have to repeat) from WATOC 2017 in Munich. Odile Eisenstein gave a talk about predicted 13 C chemical shifts in transition metal (and often transient) complexes, with the focus on metallacyclobutanes. These calculations include full spin-orbit/relativistic corrections, essential when the carbon is attached to an even slightly relativistic element.

The iron complex shown below forms the basis for many catalysts.[cite]10.1002/anie.200502985[/cite] With iron, the catalytic behaviour very much depends on the spin-state of the molecule, which for the below can be either high (hextet) or medium (quartet) spin, with a possibility also of a low spin (doublet) state. Here I explore whether structural information in crystal structures can reflect such spin states.

Conformational polymorphism occurs when a compound crystallises in two polymorphs differing only in the relative orientations of flexible groups ( e.g. Ritonavir).[cite]10.1039/D1SC06074K[/cite] At the Beilstein conference, Ian Bruno mentioned another type; tautomeric polymorphism , where a compound can crystallise in two forms differing in the position of acidic protons. Here I explore three such examples.
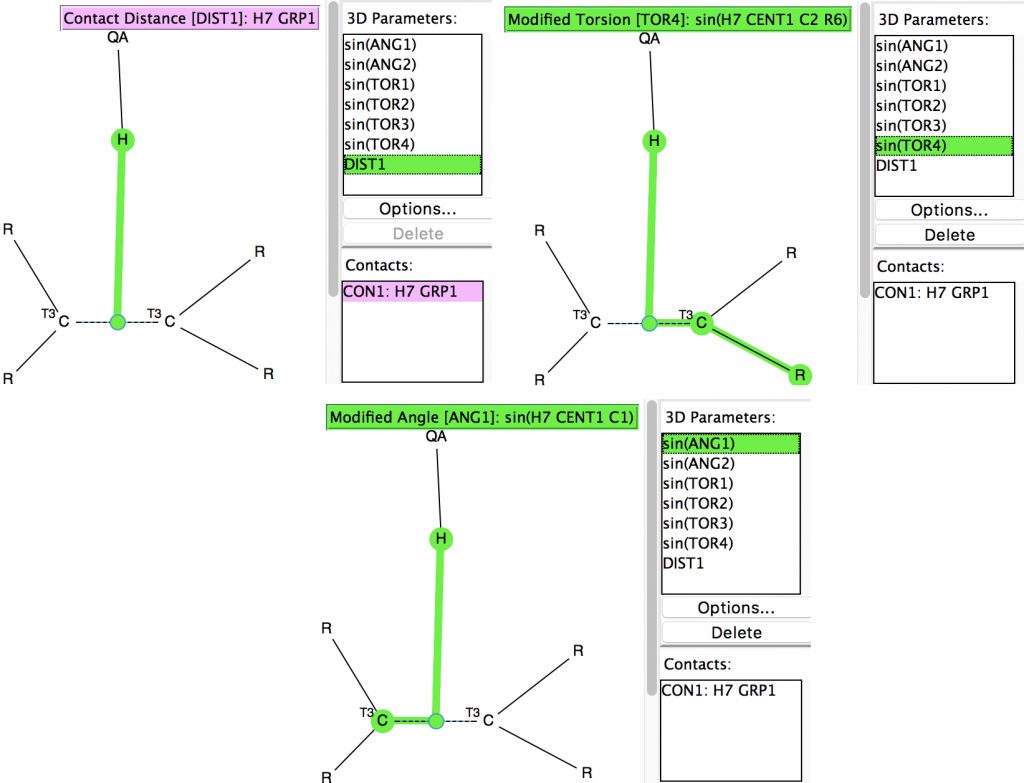
Back in the early 1990s, we first discovered the delights of searching crystal structures for unusual bonding features.[cite]10.1039/P29940000703[/cite] One of the first cases was a search for hydrogen bonds formed to the π-faces of alkenes and alkynes. In those days the CSD database of crystal structures was a lot smaller (<80,000 structures; it’s now ten times larger) and the search software less powerful.
Layer stacking in structures such as graphite is well-studied. The separation between the π-π planes is ~3.35Å, which is close to twice the estimated van der Waals (vdW) radius of carbon (1.7Å). But how much closer could such layers get, given that many other types of relatively weak interaction such as hydrogen bonding can contract the vdW distance sum by up to ~0.8Å or even more?

Following my conformational exploration of enols, here is one about a much more common molecule, a carboxylic acid. The components of the search are shown as four queries below, which will be combined in various Boolean senses (DOI: 10.14469/hpc/2462). Query one defines the carboxylic acid, with 3-coordinate carbon specified at the carbonyl along with 1-coordinate for the carbonyl oxygen.