The SN1 Reaction- revisited

In an earlier post I wrote about the iconic SN1 solvolysis reaction, and presented a model for the transition state involving 13 water molecules. Here, I follow this up with an improved molecule containing 16 water molecules, and how the barrier for this model compares with experiment. This latter is nicely summarized in the following article: Solvolysis of t-butyl chloride in water-rich methanol + water mixtures, which (for pure water) cites the following activation parameters
- ΔH†283 = 23.0 kcal/mol
- ΔG†283 = 19.7 kcal/mol
- ΔS†283 = +11.1 cal/mol/K
But first, a word about how this new transtion state has been obtained. The DFT treatment used is quite standard (B3LYP/6-31G(d) ), and one can indeed locate a transition state using just this approach (this is how the previous model was obtained). One has to work very hard to orient the starting guess for the geometry so that as many hydrogen bonds between the waters themselves, and to the substrate, are created. The previous model took quite a few guesses and attempts! The solvent in such a model is simulated by the explicit water molecules themselves. Of course, the quality of the solvent then depends on how many water molecules are used. A proper solvent field using explicit water molecules is thought to require 100s of water molecules! But a reasonable approximation/compromise may well be 13.
So how can the model be improved? Well, in many ways, some of which include treating the dynamics of the system. But I will stick just to two.
- Firstly, we assume that the water molecules are used to form a bridge between the incoming nucleophile (another water) and the leaving group (the chloride). In the previous model, two such bridges were constructed using the 13 water molecules. But in fact, there is still space between two of the methyl groups to construct a third bridge. This takes the total solvent molecules to 16.
- Solvent can also be modelled as a continuum, in which a cavity which the substrate occupies is surrounded by a field generated by the continuum solvent. The problem with these cavity approaches in the past has been that it is not easy to optimize the geometry of the molecule contained within the cavity. Because the cavity was constructed by tesselation, the first derivatives of the energy of the molecule within the cavity were not regular, and as a result, geometry optimization (and particularly transition state optimization) would frequently meander and fail to converge. Darrin York and Martin Karplus came to the rescue (some time ago, it has to be said, DOI: 10.1021/jp992097l) by formulating a smoothed out solvation cavity where the first (and second derivatives) are stable and well behaved. This new algorithm has now been implemented in Gaussian09, and it now allows really easy transition state location within a solvent cavity
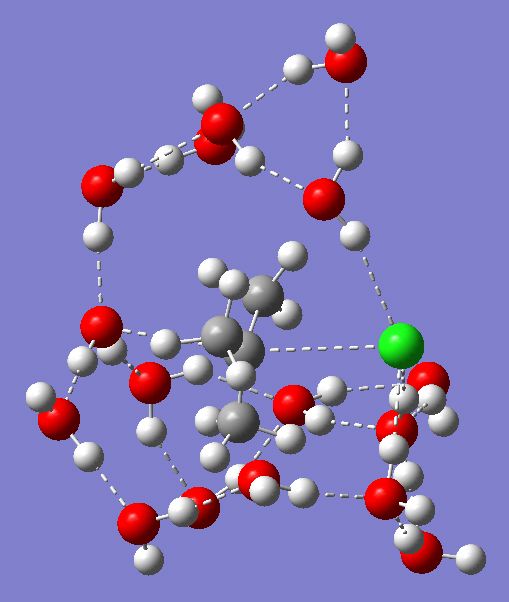
The result of this optimization is shown below (and can be seen in original form at the following DOI: 10042/to-2894).
Transition state for Sn1 solvolysis of tert-butyl chloride. Click for animation.
The model has not changed that much compared to before. The reaction (imaginary) mode still clearly shows formation of the C-O bond and cleavage of the C-Cl bond. Also as before, there is a lot of motion of the methyl groups, as the forming cation induces stereo-electronic alignment with the adjacent C-H bonds (and which explains the large secondary deuterium isotope effects measured for this reaction, kH/kD (298) = 2.39, see DOI: 10.1021/ja01080a004). The hydrogen bonding pattern is also retained (despite the surrounding solvent field!). But what of the predicted activation parameters
- ΔH†298 = 17.4 kcal/mol
- ΔG†298 = 18.7 kcal/mol
- ΔS†298 = -4.4 cal/mol/K
The overall free energy is in great agreement with experiment! But the entropy is the wrong sign!! The calculation is predicting that the transition state is more rigid than the reactant. One can see how this might happen, since the greater ionic character produces very much stronger hydrogen bonds, which strengthen the three solvent bridges. It may be simply that the rigid-rotor-harmonic-oscillator approximation breaks down horribly for the entropy in this calculation. But it is encouraging that the activation barrier is reproducing experiment, which suggests the model cannot be completely wrong!
Additional details
Description
In an earlier post I wrote about the iconic S N 1 solvolysis reaction, and presented a model for the transition state involving 13 water molecules. Here, I follow this up with an improved molecule containing 16 water molecules, and how the barrier for this model compares with experiment.
Identifiers
- UUID
- 22caa41a-7e23-44ae-88ab-4dc9927018ec
- GUID
- http://www.ch.ic.ac.uk/rzepa/blog/?p=1135
- URL
- https://www.ch.imperial.ac.uk/rzepa/blog/?p=1135
Dates
- Issued
-
2009-11-11T10:48:36
- Updated
-
2011-06-12T11:16:42