Veröffentlicht in Henry Rzepa's Blog
Autor Henry Rzepa
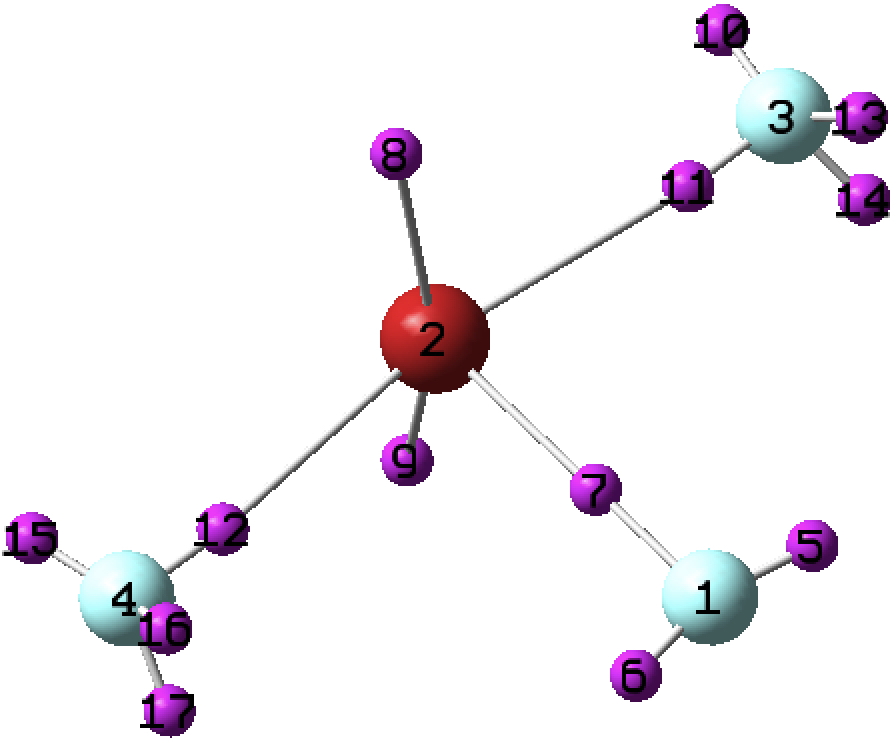
I analysed the bonding in chlorine trifluoride a few years back in terms of VSEPR theory. I noticed that several searches on this topic which led people to this post also included a query about the differences between it and the bromine analogue. For those who posed this question, here is an equivalent analysis.