
In Vivo Base Editing of PCSK9: Early Clinical Data from VERVE-102 and the Path Toward Durable LDL-C Control
Creators & Contributors

The interim analysis of the Heart-2 trial of VERVE-102 provides one of the clearest public demonstrations to date that in vivo adenine base editing can produce substantial and durable pharmacodynamic reductions in circulating PCSK9 protein and LDL cholesterol following a single intravenous infusion. [1] The importance of these findings does not stem from PCSK9 as a therapeutic target. That biology has been validated through human genetics and multiple pharmacologic modalities for more than two decades [3–8]. What is new is the demonstration that a somatic genomic edit intended to persist in edited hepatocytes can achieve sustained suppression of a validated cardiovascular risk pathway in humans.
The open-label, single-ascending-dose Phase 1b study enrolled 35 adults with heterozygous familial hypercholesterolemia or premature coronary artery disease who required additional LDL-C lowering despite maximally tolerated oral therapy. Participants received VERVE-102 across six dose cohorts ranging from 0.3 to 1.0 mg total RNA per kilogram of body weight. As of the February 2026 data cutoff, the median follow-up was approximately nine months, with 15 participants observed for at least one year and some reaching 18 months. All participants are expected to enroll in a long-term safety registry extending to 15 years. [1]
Mean PCSK9 reductions increased from 51% at the lowest dose to 88% at the highest dose. Corresponding mean LDL-C reductions were 9%, 44%, 45%, 33%, 51%, and 62% across the ascending cohorts, with an absolute reduction of 78 mg/dL at the 1.0 mg/kg dose. Both analytes exhibited durable suppression over the available observation period. [1] These data extend the earlier interim report from the first three cohorts (14 participants) reported in April 2025.
Table 1. Mean reductions in LDL cholesterol by dose cohort in the Heart-2 interim analysis (Vafai et al., N. Engl. J. Med., 2026). [1]
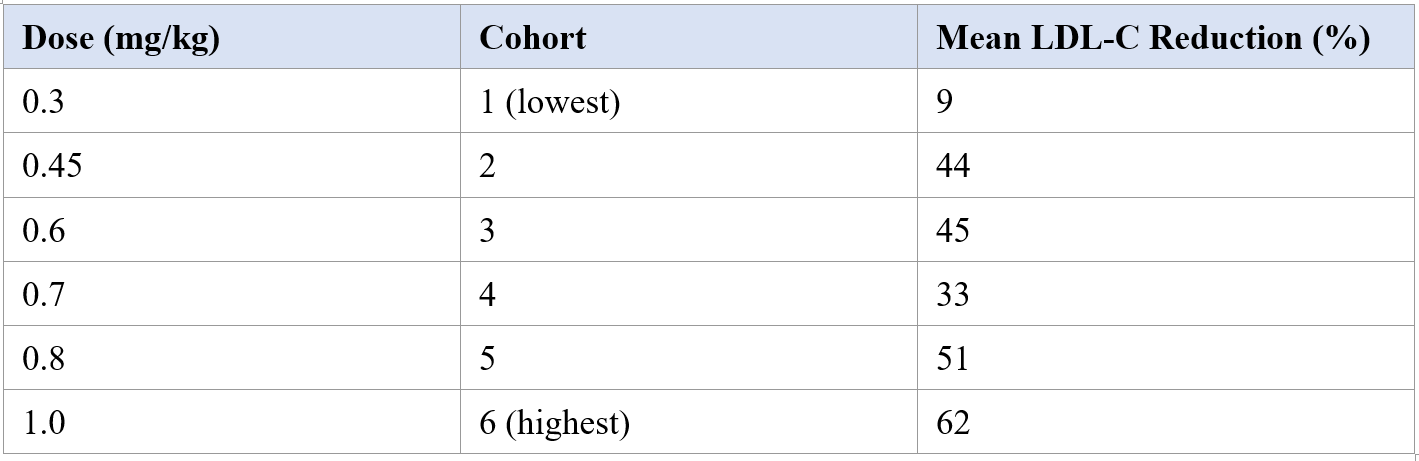
The non-monotonic LDL-C response at the 0.7 mg/kg cohort (33% mean reduction) deviates from the otherwise generally dose-dependent pattern. With only a small number of participants per cohort, this departure is most reasonably interpreted as random variability rather than a true pharmacodynamic inflection point. Per-cohort estimates from early-phase trials of this size carry wide confidence intervals, and overinterpretation of any individual dose level prior to adequately powered studies would be premature.
These pharmacodynamic signals are substantial. They are also scientifically demanding in a manner that distinguishes this modality from reversible pharmacologic approaches. A monoclonal antibody, small interfering RNA, or small-molecule inhibitor can be titrated, withheld, or discontinued. A successful in vivo base edit is intended to persist in edited hepatocytes and their progeny. That irreversibility fundamentally alters the evidentiary requirements. For VERVE-102, the central question is not merely whether LDL-C falls, but whether the analytical, genomic, manufacturing, and long-term clinical evidence is sufficient to justify an indelible edit in a prevalent cardiovascular-risk population.
VERVE-102 has received FDA Fast Track designation for the treatment of hyperlipidemia and high lifetime cardiovascular risk. [2] Fast Track status reflects regulatory recognition that the program addresses a serious condition and may meet an unmet medical need; it does not alter the evidentiary threshold for approval.
Genetic Validation and Therapeutic Context
The causal relationship between PCSK9 activity, LDL receptor turnover, and circulating LDL-C was established through human genetics well before any pharmacologic inhibitor reached the clinic. Individuals carrying PCSK9 loss-of-function variants exhibit lifelong reductions in LDL-C, with the magnitude depending on the specific variant and zygosity, and are correspondingly protected from atherosclerotic cardiovascular events. [3,4] These observations supplied both target validation and a natural benchmark: any successful therapy should ideally reproduce the magnitude and durability of protection conferred by these natural genetic models without introducing novel liabilities.
Current approved modalities achieve substantial LDL-C lowering but remain imperfect in practice. Statin meta-analyses demonstrate that each 1 mmol/L reduction in LDL-C is associated with a proportional reduction in major vascular events. [5] PCSK9-directed monoclonal antibodies, evolocumab and alirocumab, each produced significant cardiovascular outcome benefits in adequately powered phase 3 trials. [6,7] Inclisiran, a small interfering RNA administered subcutaneously twice yearly, demonstrated sustained LDL-C lowering in two phase 3 programs. [8] Yet real-world adherence to these regimens is variable, and many patients with HeFH or established atherosclerotic disease remain above guideline-recommended LDL-C targets. A single intervention capable of producing comparable or greater LDL-C reduction with multi-year or lifelong durability would address a recognized unmet need in adherence and convenience, provided its safety and genomic specificity are supported by evidence strong enough for regulatory review and clinical adoption.
Mechanism and Trial Design
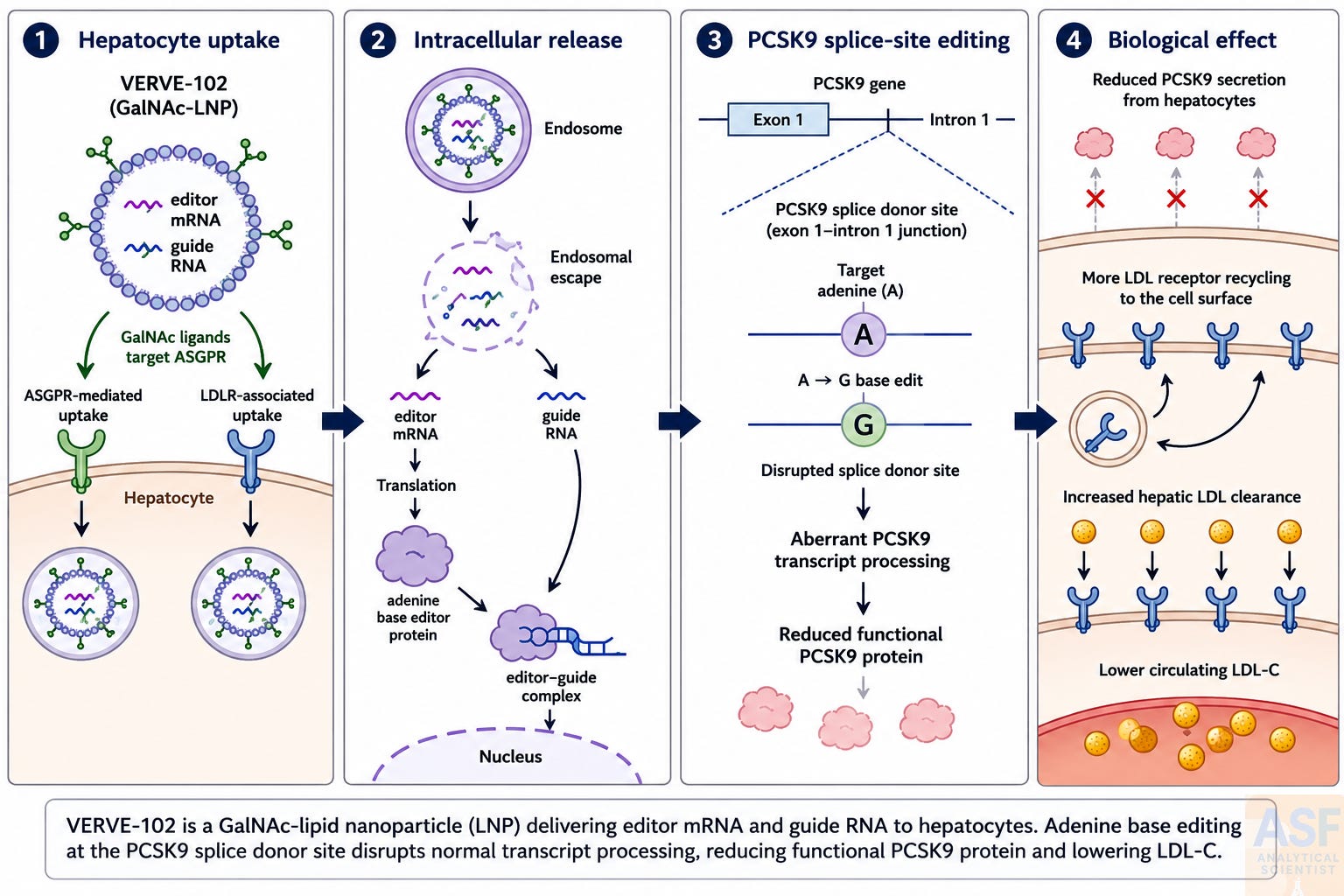
VERVE-102 consists of two RNA drug substances: messenger RNA encoding an adenine base-editor protein and a guide RNA targeting PCSK9. These are co-encapsulated in a GalNAc-lipid nanoparticle. Hepatocyte uptake occurs via endogenous LDL receptor-mediated internalization and GalNAc-mediated endocytosis via the asialoglycoprotein receptor. The dual mechanism is clinically relevant because ASGPR-mediated delivery is independent of LDLR expression and may remain effective even in patients with substantial LDLR deficiency.
Once internalized, the mRNA is translated transiently into the adenine base-editor protein, a fusion of an engineered adenosine deaminase with a catalytically impaired CRISPR-Cas9 nickase. The guide RNA directs this complex to the splice donor site at the junction of exon 1 and intron 1 of the PCSK9 gene. There, the adenosine deaminase catalyzes an A-to-G nucleotide conversion that disrupts the canonical GT splice donor consensus sequence. The resulting aberrant transcript processing is expected to reduce production of functional PCSK9 protein. Because the edit is written into genomic DNA, the effect is expected to persist in edited hepatocytes and their progeny.
Adenine base editing enables A·T-to-G·C conversion without double-strand DNA cleavage [9]. A closely related in vivo PCSK9 base-editing strategy was later shown to produce durable PCSK9 knockdown and LDL-C reduction in cynomolgus macaques following a single LNP infusion [10]. VERVE-102 employs the same PCSK9 guide RNA and a similar adenine base-editor mRNA as the earlier VERVE-101 formulation but uses a different ionizable lipid and incorporates the GalNAc liver-targeting ligand in a distinct LNP architecture. [11]
Heart-2 is an open-label, single-ascending-dose Phase 1b study (ClinicalTrials.gov identifier NCT06164730) conducted in adults with HeFH or premature coronary artery disease who required additional LDL-C lowering despite maximally tolerated oral lipid-lowering therapy. [1] Thirty-five participants across six dose cohorts received a single intravenous infusion and were followed for a minimum of 28 days.
Efficacy Observations
The primary pharmacodynamic signals were robust and generally dose-dependent. Mean PCSK9 reductions increased from 51% at 0.3 mg/kg to 88% at 1.0 mg/kg. Corresponding mean LDL-C reductions are summarized in Table 1, with an absolute reduction of 78 mg/dL at the highest dose. Both analytes showed durable suppression over the available observation period. [1]
Safety Observations
Early tolerability was encouraging, but the dataset is too small and too short to characterize the long-term safety profile of an in vivo editing intervention intended to persist in edited hepatocytes. No dose-limiting toxicities and no treatment-related serious adverse events were reported. All 35 participants received their full planned dose and none withdrew for safety-related reasons. [1] Adverse events attributed to VERVE-102 consisted primarily of mild-to-moderate infusion-related reactions and fatigue. Transient ALT elevations were observed but were not accompanied by clinically significant changes in other liver function parameters or progression to severe hepatotoxicity. One participant experienced aspiration pneumonitis in the setting of pre-existing gastroesophageal reflux disease; a causal relationship to study drug was not established.
These findings should be interpreted against the backdrop of the earlier VERVE-101 (Heart-1) program. In April 2024, enrollment in the Heart-1 trial was halted after a patient in the 0.45 mg/kg cohort developed Grade 3 ALT elevation and Grade 3 thrombocytopenia within four days of dosing. [11] The events were interpreted as likely related to the LNP delivery system rather than to the base-editing mechanism itself. The absence of corresponding laboratory abnormalities in the Heart-2 dataset through 35 participants is encouraging, but it does not yet establish that delivery-related laboratory abnormalities have been eliminated. Nevertheless, the Heart-2 follow-up period remains substantially shorter than what will be required for a complete long-term safety characterization.
Gene therapy history is relevant here. Several programs have shown that early tolerability does not eliminate the need for larger populations and longer observation. The current dataset, while favorable in the near term, does not yet exclude risks that manifest over years, nor does it provide the quantitative genome-wide off-target editing data in human samples that will ultimately be required to characterize genotoxic risk.
Critical Appraisal: Strengths, Limitations, and Open Questions
The principal strength of the reported data lies in the clear demonstration of on-target pharmacologic activity that is dose-dependent and persists well beyond the expected duration of transient mRNA and protein expression. Durability of LDL-C reduction at 18 months in some participants supports the mechanistic premise that a single genomic base edit can produce a lasting change in hepatocyte phenotype. For a Phase 1b study conducted in a high-cardiovascular-risk population already on background lipid-lowering therapy, the absence of treatment-related serious adverse events through 35 participants across a fourfold dose range is a meaningful safety signal and justifies continued clinical development.
The limitations are not subtle. With 35 participants and a median follow-up of approximately nine months, the dataset provides proof-of-concept, not definitive evidence of clinical benefit or long-term safety. The publicly available data do not include quantitative on-target editing efficiency—the percentage of PCSK9 alleles bearing the A-to-G edit in liver biopsy or surrogate tissue—nor do they include comprehensive genome-wide off-target base-editing rates or indel frequencies from human samples. These parameters are central to the genotoxicity risk assessment for any genome-editing product and will be critical components of the regulatory evidence package. Sponsors will need to demonstrate, through validated next-generation sequencing methods and standardized bioinformatic pipelines, that off-target events at clinically meaningful frequencies are absent or acceptably rare in target tissues.
There is also the question of dose selection for Phase 2. The current data suggest that doses in the 0.8 to 1.0 mg/kg range produce the largest mean LDL-C reductions, but variability within cohorts and the non-monotonicity at 0.7 mg/kg mean that larger, statistically powered studies will be needed to define the optimal dose with confidence. The relationship between percent allele editing, PCSK9 suppression, LDL-C lowering, and ultimately cardiovascular event reduction will need to be formally modeled and validated before pharmacodynamic endpoints can serve as reliable surrogates for outcome endpoints in pivotal trials.
The Central Role of Analytical Science
Translation of in vivo base editors from concept to clinic depends critically on the quality and validation of analytical methods deployed across discovery, nonclinical development, manufacturing, and clinical monitoring. Critical quality attributes of the GalNAc-LNP mRNA drug product include particle size distribution and polydispersity index (assessed by dynamic light scattering or asymmetric flow field-flow fractionation with multi-angle light scattering), mRNA encapsulation efficiency and integrity (by fluorometric assay and ion-pair reversed-phase HPLC or capillary electrophoresis), GalNAc ligand density and conjugation uniformity, and residual process-related impurities including double-stranded RNA species that are potent activators of innate immune sensing pathways. Each of these attributes directly influences biodistribution, hepatocyte uptake, translation efficiency, editing precision, and immunogenicity.
A potency assay must link these physicochemical parameters to a biologically relevant readout: on-target editing rate at the PCSK9 locus by next-generation sequencing, PCSK9 protein knockdown in primary human hepatocytes, or functional upregulation of LDL receptor surface expression. This linkage anchors the release specification to clinical effect.
From a regulatory science standpoint, the International Council for Harmonisation Q2(R2) guideline on analytical procedure validation applies directly to this product class. [12] Release testing, stability studies, and comparability assessments across manufacturing scales require demonstration of accuracy, precision, specificity, linearity, range, and robustness for each analytical procedure. For the genomic safety package, the FDA has issued both a finalized guidance on human genome editing products [13] and a recent draft guidance on the specific use of NGS methods for off-target safety assessment. [14] That draft guidance provides recommendations on NGS-based methods, study design, sample selection, assay sensitivity, bioinformatic pipelines, and reporting of off-target editing assessments in nonclinical studies supporting investigational genome-editing products [14].
The transient ALT elevations observed in Heart-2, while not dose-limiting, underscore the importance of sensitive multiplexed biomarker panels that can distinguish innate immune activation from direct hepatotoxicity. From an analytical perspective, the VERVE-102 clinical claim depends on an unbroken measurement chain: LNP composition and critical quality attributes link to liver delivery and editing activity, which link to PCSK9 suppression, which links to LDL-C lowering, which is expected to link to cardiovascular risk reduction. Any break in that measurement chain could compromise both the safety evidence package and the consistency of the commercial product. The analytical control strategy is therefore not peripheral to VERVE-102. It is part of the evidence needed to make the clinical claim credible.
Implications for Development, Regulation, and Clinical Practice
If subsequent adequately powered trials confirm durable LDL-C reduction with an acceptable long-term safety and genomic specificity profile, VERVE-102 or mechanistically related agents could occupy a well-defined clinical niche: patients with HeFH who have not reached LDL-C goal on maximal oral therapy, or individuals with established atherosclerotic cardiovascular disease who face decades of adherence burden with injectable biologics or twice-yearly siRNA. Treatment guidelines would require updating to incorporate genomic intervention options, with explicit criteria for patient selection, pre-treatment counseling on the intended durability and irreversibility of somatic editing, and protocols for long-term genomic surveillance.
The regulatory pathway for the first in vivo base editor indicated for a prevalent cardiovascular condition will establish important precedents. Sponsors will need to satisfy heightened expectations for off-target characterization, on-target editing efficiency documentation, manufacturing consistency under current Good Manufacturing Practice for human gene therapy and genome-editing products, and long-term follow-up commitments (already planned at 15 years for Heart-2 participants). The FDA April 2026 draft guidance on NGS-based safety assessment provides a concrete framework. [14] Post-marketing commitments will likely include pharmacovigilance programs designed to detect genotoxicity signals, including off-target edits, indels, chromosomal rearrangements, clonal expansion, or oncogenic selection in patient populations followed over decades.
Access will shape the clinical impact of any approved one-time editing therapy. Experience with approved one-time genetic medicines suggests that pricing, reimbursement frameworks, upfront budget impact, and payer willingness to recognize long-term value will shape patient access as much as efficacy data. Early engagement with patient communities, health technology assessment bodies, and payer stakeholders during the clinical development phase is advisable if the aspiration is to make durable LDL-C control broadly available.
Scientifically, the path forward requires Phase 2 data in larger, more diverse populations with pre-specified cardiovascular outcome endpoints or validated surrogate endpoints linked to hard clinical events. Publicly reported on-target editing efficiencies from liver biopsy or validated surrogate measurements, together with genome-wide off-target profiling using orthogonal NGS-based approaches, will be necessary to establish the complete benefit-risk profile. Continued investment in validated analytical methods for potency, LNP critical quality attributes, and sensitive off-target detection, aligned with ICH Q2(R2) [12] and the emerging FDA regulatory expectations described in the 2024 and 2026 guidance documents [13,14], remains foundational to that evidence generation.
Conclusion
The VERVE-102 Heart-2 interim data represent a meaningful technical milestone: among the clearest public demonstrations to date that in vivo base editing can produce durable pharmacodynamic modulation of a major cardiovascular risk factor following a single administration. The target is genetically well validated, and the Heart-2 data now provide human pharmacodynamic proof of concept over the available follow-up. The results merit accelerated but disciplined investigation.
The history of gene therapy cautions that early-phase enthusiasm has periodically required subsequent recalibration when safety signals emerged in larger populations. Contemporary base editors offer improved molecular specificity relative to earlier nuclease-based approaches, yet the principles of rigorous evidence generation, analytical transparency, and candid communication of uncertainty remain indispensable.
Whether this approach ultimately changes cardiovascular prevention will be determined less by the elegance of its molecular mechanism than by the completeness and quality of the evidence that follows. Larger and more diverse studies with pre-specified outcome endpoints, comprehensive public reporting of on-target editing efficiencies and genome-wide off-target profiles, and sustained investment in validated analytical methods for potency and genomic safety assessment will be required to convert a promising pharmacodynamic signal into reliable clinical confidence. If those standards are met with the necessary rigor and transparency, the prospect of moving selected patients from lifelong pharmacologic management toward a durable, one-time genomic intervention for a common cardiovascular risk factor becomes genuinely tangible. Realizing that prospect will require evidence generation as disciplined as the molecular intervention itself.
References
[1] S. B. Vafai et al., "In vivo base editing of PCSK9 with VERVE-102 for hypercholesterolemia," N. Engl. J. Med., May 2026. https://doi.org/10.1056/NEJMoa2601283
[2] Verve Therapeutics, "Verve Therapeutics receives U.S. FDA Fast Track designation for VERVE-102, an in vivo base editing medicine targeting PCSK9," Apr. 11, 2025. [Online]. Available: https://vervetx.gcs-web.com/news-releases/news-release-details/verve-therapeutics-receives-us-fda-fast-track-designation-verve/
[3] J. C. Cohen, E. Boerwinkle, T. H. Mosley Jr., and H. H. Hobbs, "Sequence variations in PCSK9, low LDL, and protection against coronary heart disease," N. Engl. J. Med., vol. 354, no. 12, pp. 1264–1272, Mar. 2006. https://doi.org/10.1056/NEJMoa054013
[4] J. Cohen et al., "Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9," Nat. Genet., vol. 37, no. 2, pp. 161–165, Feb. 2005. https://doi.org/10.1038/ng1509
[5] Cholesterol Treatment Trialists' Collaboration, "Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials," Lancet, vol. 376, no. 9753, pp. 1670–1681, Nov. 2010. https://doi.org/10.1016/S0140-6736(10)61350-5
[6] M. S. Sabatine et al., "Evolocumab and clinical outcomes in patients with cardiovascular disease," N. Engl. J. Med., vol. 376, no. 18, pp. 1713–1722, May 2017. https://doi.org/10.1056/NEJMoa1615664
[7] G. G. Schwartz et al., "Alirocumab and cardiovascular outcomes after acute coronary syndrome," N. Engl. J. Med., vol. 379, no. 22, pp. 2097–2107, Nov. 2018. https://doi.org/10.1056/NEJMoa1801174
[8] K. K. Ray et al., "Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol," N. Engl. J. Med., vol. 382, no. 16, pp. 1507–1519, Apr. 2020. https://doi.org/10.1056/NEJMoa1912387
[9] N. M. Gaudelli et al., "Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage," Nature, vol. 551, no. 7681, pp. 464–471, Nov. 2017. https://doi.org/10.1038/nature24644
[10] K. Musunuru et al., "In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates," Nature, vol. 593, no. 7859, pp. 429–434, May 2021. https://doi.org/10.1038/s41586-021-03534-y
[11] Verve Therapeutics, "Verve Therapeutics announces updates to its PCSK9 program," Apr. 2, 2024. [Online]. Available: https://vervetx.gcs-web.com/news-releases/news-release-details/verve-therapeutics-announces-updates-its-pcsk9-program/
[12] International Council for Harmonisation, Validation of Analytical Procedures Q2(R2), Final version, Nov. 2023. [Online]. Available: https://database.ich.org/sites/default/files/ICH_Q2%28R2%29_Guideline_2023_1130.pdf
[13] U.S. Food and Drug Administration, Human Gene Therapy Products Incorporating Human Genome Editing: Guidance for Industry. Silver Spring, MD: Center for Biologics Evaluation and Research, Jan. 2024. [Online]. Available: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-products-incorporating-human-genome-editing
[14] U.S. Food and Drug Administration, Safety Assessment of Genome Editing in Human Gene Therapy Products Using Next-Generation Sequencing: Draft Guidance for Industry. Silver Spring, MD: Center for Biologics Evaluation and Research, Apr. 2026. Docket no. FDA-2026-D-1255. [Online]. Available: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/safety-assessment-genome-editing-human-gene-therapy-products-using-next-generation-sequencing
Additional details
Description
Phase 1b data from Heart-2 show sustained PCSK9 suppression and LDL-C lowering after a single genomic intervention
Identifiers
- GUID
- 199663564
- URL
- https://abrahamfinny.substack.com/p/in-vivo-base-editing-of-pcsk9-early
Dates
- Issued
-
2026-06-01T13:04:17
- Updated
-
2026-06-01T13:04:17
References
- Vafai, S. B., Täubel, J., Ashdown, T., Patel, R. S., Diamondali, S., Cegla, J., Soran, H., Bashir, B., Abitbol, A., Gaudet, D., Lauzière, A., Brunham, L. R., Newby, D. E., Nicholls, S. J., Scott, R. S., Kerr, J., Tardif, J.-C., Lunken, C., Humphries, S. E., … Kathiresan, S. (2026). In Vivo Base Editing of PCSK9 with VERVE-102 for Hypercholesterolemia. New England Journal of Medicine. https://doi.org/10.1056/nejmoa2601283
- Unknown title https://vervetx.gcs-web.com/news-releases/news-release-details/verve-therapeutics-receives-us-fda-fast-track-designation-verve/
- Cohen, J. C., Boerwinkle, E., Mosley, T. H., & Hobbs, H. H. (2006). Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. New England Journal of Medicine, 354(12), 1264–1272. https://doi.org/10.1056/nejmoa054013
- Cohen, J., Pertsemlidis, A., Kotowski, I. K., Graham, R., Garcia, C. K., & Hobbs, H. H. (2005). Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nature Genetics, 37(2), 161–165. https://doi.org/10.1038/ng1509
- Cholesterol Treatment Trialists' (CTT) Collaboration. (2010). Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. The Lancet, 376(9753), 1670–1681. https://doi.org/10.1016/s0140-6736(10)61350-5
- Sabatine, M. S., Giugliano, R. P., Keech, A. C., Honarpour, N., Wiviott, S. D., Murphy, S. A., Kuder, J. F., Wang, H., Liu, T., Wasserman, S. M., Sever, P. S., & Pedersen, T. R. (2017). Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. New England Journal of Medicine, 376(18), 1713–1722. https://doi.org/10.1056/nejmoa1615664
- Schwartz, G. G., Steg, P. G., Szarek, M., Bhatt, D. L., Bittner, V. A., Diaz, R., Edelberg, J. M., Goodman, S. G., Hanotin, C., Harrington, R. A., Jukema, J. W., Lecorps, G., Mahaffey, K. W., Moryusef, A., Pordy, R., Quintero, K., Roe, M. T., Sasiela, W. J., Tamby, J.-F., … Zeiher, A. M. (2018). Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. New England Journal of Medicine, 379(22), 2097–2107. https://doi.org/10.1056/nejmoa1801174
- Ray, K. K., Wright, R. S., Kallend, D., Koenig, W., Leiter, L. A., Raal, F. J., Bisch, J. A., Richardson, T., Jaros, M., Wijngaard, P. L. J., & Kastelein, J. J. P. (2020). Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. New England Journal of Medicine, 382(16), 1507–1519. https://doi.org/10.1056/nejmoa1912387
- Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., & Liu, D. R. (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature, 551(7681), 464–471. https://doi.org/10.1038/nature24644
- Musunuru, K., Chadwick, A. C., Mizoguchi, T., Garcia, S. P., DeNizio, J. E., Reiss, C. W., Wang, K., Iyer, S., Dutta, C., Clendaniel, V., Amaonye, M., Beach, A., Berth, K., Biswas, S., Braun, M. C., Chen, H.-M., Colace, T. V., Ganey, J. D., Gangopadhyay, S. A., … Kathiresan, S. (2021). In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature, 593(7859), 429–434. https://doi.org/10.1038/s41586-021-03534-y
- Unknown title https://vervetx.gcs-web.com/news-releases/news-release-details/verve-therapeutics-announces-updates-its-pcsk9-program/
- Unknown title https://database.ich.org/sites/default/files/ich_q2(r2)_guideline_2023_1130.pdf
- Unknown title https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-products-incorporating-human-genome-editing
- Unknown title https://www.fda.gov/regulatory-information/search-fda-guidance-documents/safety-assessment-genome-editing-human-gene-therapy-products-using-next-generation-sequencing