
FDA's 2026 CGT CMC Flexibility Guidance: Making Evidence Credible Under Constraint
Creators & Contributors


The U.S. Food and Drug Administration's (FDA) May 2026 guidance on chemistry, manufacturing, and controls (CMC) for cellular and gene therapy products clarifies how sponsors can use scientifically justified flexibility when preparing biologics license applications. Rather than lowering standards, the guidance offers meaningful flexibility in areas such as analytical method validation, process performance qualification, specifications, comparability, and stability, provided sponsors can show that the available evidence is adequate for the specific product, risk, and regulatory decision. The guidance does not support weaker evidence. It requires a clearer explanation of why the available evidence is scientifically adequate: define the constraint, identify the quality risk it creates, show why the evidence addresses that risk, and explain how remaining uncertainty will be reduced, monitored, or resolved through lifecycle controls. For many programs, success will depend less on the volume of data generated and more on the quality of the scientific reasoning that links limited data to product understanding, process control, and patient safety. This article examines what the guidance actually demands and provides a practical framework for building arguments that can withstand regulatory scrutiny.
The guidance is about making evidence credible under constraint
The central point of the guidance published on May 5th, 2026, from the U.S. Food and Drug Administration is not flexibility alone. It is flexibility without lowering the licensure standard: sponsors of human cellular and gene therapy (CGT) products must demonstrate that their products meet applicable requirements to ensure they are safe, pure, and potent [1]. The guidance concerns CMC, but its deeper message is about evidence. When conventional evidence packages are limited by small lots, short shelf life, patient-specific manufacturing, limited patient populations, or limited process history, flexibility depends on whether the sponsor can make a credible scientific case.
That case is not analytical alone. It also depends on process understanding, risk management, manufacturing controls, and lifecycle learning. Analytical science is central because it translates those elements into measurable evidence: what is known, what is controlled, what remains uncertain, and which uncertainties matter for the product being licensed under a biologics license application (BLA). FDA states that the extent of CMC data needed, and the suitability of a CMC flexibility, may vary depending on development stage, manufacturing process, and product, and that the Agency may seek additional data on a case-by-case basis [1]. In practical terms, CMC flexibility does not reduce the need for evidence. It changes how evidence must be assembled, justified, and connected to risk.
Why conventional CMC evidence models strain under CGT conditions
FDA identifies the practical pressures clearly: product complexity, patient-specific manufacturing, sophisticated processes, advanced technologies, limited patient populations, fewer manufacturing runs, product characterization and analytical testing challenges, and short product shelf life with a narrow window from production to administration [1]. Those pressures are not merely operational. They change what a persuasive evidence package can realistically look like. A conventional biologic may benefit from repeated campaigns, larger lot sizes, broader stability inventories, and enough material to test, retest, trend, and investigate. Many CGT products do not provide that margin.
An autologous cell therapy may begin with variable patient material. A viral vector product may require integration of particle or genome titer, functional activity, infectivity, impurity, identity, and potency evidence. A genome-editing product may require methods that connect molecular modification, cell phenotype, and intended biological function, without pretending that any single assay can answer every question. Critical nucleic acid or vector-related components can also create quality risks that must be understood before they become later uncertainty in clinical interpretation or manufacturing control.
FDA's 2020 human gene therapy CMC guidance illustrates the breadth of the field by including, among examples of gene therapy products, plasmids, in vitro transcribed ribonucleic acid (RNA), genetically modified microorganisms, engineered site-specific nucleases, and ex vivo genetically modified human cells [2]. The May 2026 guidance sits atop that complexity. It does not make product understanding optional. It makes it harder to conceal a lack of product understanding.
Constraint, risk, evidence, closure
A strong flexibility argument has four parts. First, name the constraint. Second, name the quality risk that the conventional evidence package would normally address. Third, identify the analytical, process, and control evidence that addresses that risk in the current submission. Fourth, state how the remaining uncertainty will be reduced, monitored, or closed through continued process verification, post-approval specification reevaluation, stability follow-up, or post-approval monitoring of commercial batch data. This is more than a discipline of submission writing. It is a way to prevent the most common failure mode in flexible CMC arguments: treating practical difficulty as if it were scientific justification. Limited material, few lots, or a narrow release window may explain why a conventional package is difficult to achieve. They do not, by themselves, show that the alternative package is adequate.
A practical example: single-lot validation is not single-source evidence
Consider a patient-specific autologous T-cell therapy nearing BLA submission. The manufacturing process is defined, but each lot begins with a different patient leukapheresis material. The final product is limited and clinically consequential. The release window is narrow. Repeating a full conventional validation design across multiple final-product lots would consume material that may be needed for treatment or essential quality testing. The sponsor proposes to validate a release method using one representative commercial-process lot. FDA says it may consider analytical method validation using a single representative lot when scientifically supported and when the data show that the method is sufficiently robust and suitable for its intended use [1].
A weak argument would stop at feasibility: this is a rare product, the sample volume is limited, and additional lots are hard to obtain. Those statements describe the problem. They do not solve it. The stronger argument starts with representativeness. Why does this lot reflect the commercial process? What patient-material variability has already been observed? Which method parameters were challenged? Were precision, specificity, matrix effects, reportable range, and robustness assessed in a way that matches the release decision? How do acceptance criteria relate to clinical manufacturing experience? Which performance signals will be trended as commercial lots accumulate?
In that version, the single lot is not treated as the whole evidence package. It is one part of a larger argument that includes prior clinical manufacturing data, process controls, in-process measurements, orthogonal characterization where appropriate, and lifecycle verification. That is the difference between asking FDA to accept a smaller package and showing why the available package is scientifically meaningful.
Each flexibility has an evidentiary counterpart
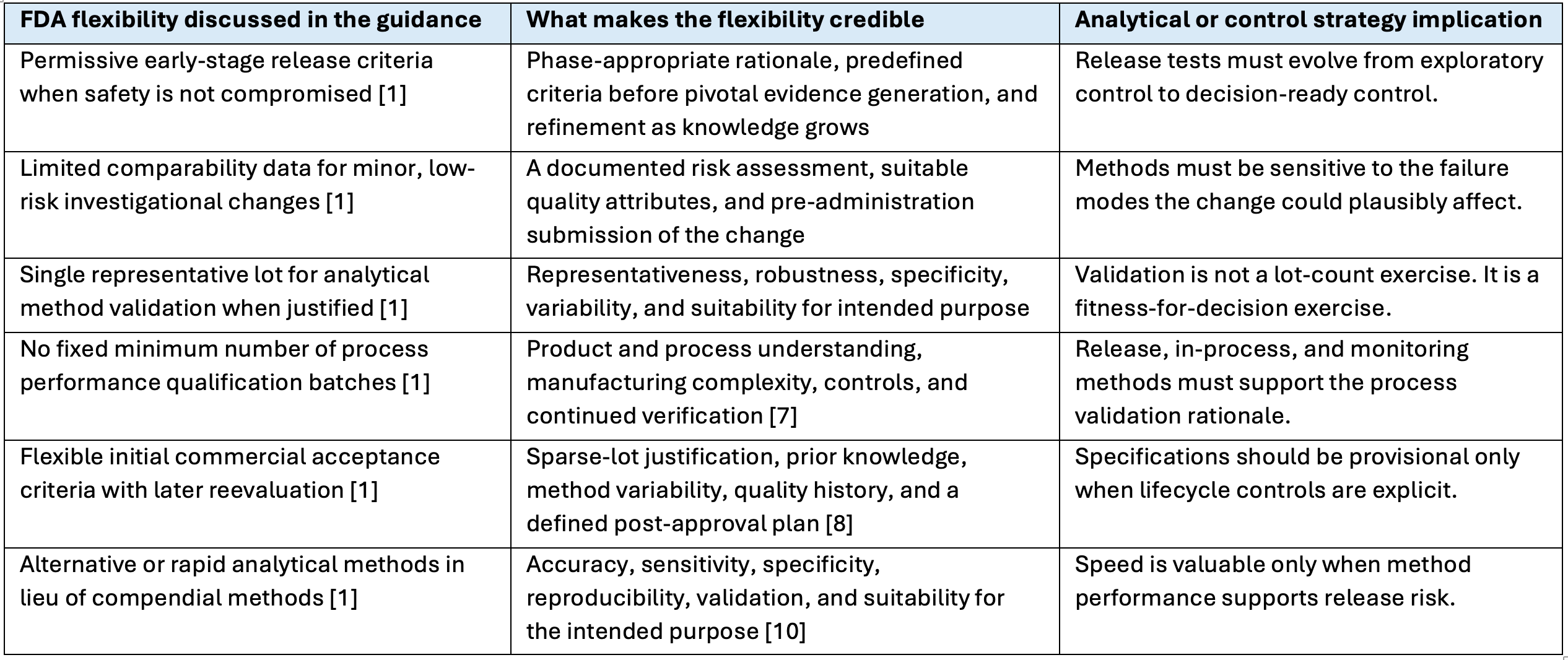
The guidance is most useful when read as a set of conditional flexibilities rather than a menu of shortcuts. Each flexibility has an evidentiary counterpart.
Table 1. FDA CMC Flexibilities and the Evidence Needed to Make Them Credible
The analytical target profile should anchor the argument
International Council for Harmonisation (ICH) Q14 Analytical Procedure Development gives the field useful language for this problem. It describes science- and risk-based analytical procedure development and defines the analytical target profile as a prospective summary of the intended purpose of an analytical procedure, the product attributes to be measured, and the relevant performance characteristics with associated criteria [3]. For CGT products, the analytical target profile matters because method choice is inseparable from biological significance, sample limitation, manufacturing risk, and release timing.
Different decisions require different methods. A release method may need speed, robustness, clear acceptance criteria, and compatibility with limited sample volume. A characterization method may need deeper mechanistic information, even if it is not suitable for routine release. A comparability method may need sensitivity to process-related changes that alter product quality. A stability-indicating method must detect degradation or loss of function that is relevant to product quality, not merely a convenient signal decline.
ICH Q2(R2) Validation of Analytical Procedures reinforces this point by framing validation around the ability of a procedure to be fit for its intended purpose and by describing performance characteristics such as specificity, accuracy, precision, detection limit, quantitation limit, range, and robustness, as appropriate to the method and use [4]. In CGT development, the same assay can be excellent for one decision and inadequate for another.
This distinction should be explicit in the CMC narrative. A flow cytometry method for cluster of differentiation 3-positive (CD3+) identity or cluster of differentiation 4-positive (CD4+) and cluster of differentiation 8-positive (CD8+) composition may support important cell product decisions, but it cannot, by itself, establish biological potency. A vector genome titer method may be essential, but it is not equivalent to functional transduction or expression. A residual impurity method may protect safety and process control, but it says little about the mechanism. The value of the package lies in explaining what each assay is for, what it is not for, and how the methods together support control.
Potency is where analytical science and biology meet
Potency is often the attribute that most clearly exposes the difficulty of CGT analytics. FDA's December 2023 draft guidance on potency assurance describes potency assurance as a multifaceted strategy that can include potency assays, manufacturing process controls, characterization tests, in-process tests, specifications, and stability studies [6]. That framing matters because a single release assay may not capture the full biological rationale of a complex therapy.
For a cell therapy, potency may depend on phenotype, viability, differentiation state, activation state, persistence, or functional response. For an adeno-associated virus (AAV) vector, potency may depend on transduction and expression, as well as the intended biological activity, while product quality may also depend on capsid-related attributes, residual host cell proteins, residual nucleic acids, aggregates, and other process-related impurities. For a genome-editing product, the relevant evidence may require linkage among edit, expression, phenotype, and safety-related off-target or impurity considerations. These examples are not a request for excessive testing. They are a reminder that potency assurance must align with the mechanism and risk.
The mistake is to treat potency as a release test alone rather than as part of a lifecycle potency strategy. A stronger approach builds a potency argument across the lifecycle. Early methods may be exploratory and imperfect, but they should still teach the sponsor which attributes plausibly matter. Later methods should be qualified or validated for the decisions they are expected to support. Based on the BLA review, the sponsor should be able to explain why the potency strategy is suitable for the product and how it remains aligned with manufacturing control, stability, comparability, and clinical interpretation.
Comparability is where method relevance is tested
The May 2026 guidance states that the FDA may accept limited comparability data for an investigational product in circumstances where the manufacturing change is considered minor and low risk to product quality, provided the product quality attributes are met, and the change is submitted before administering the post-change product [1]. The 2023 draft guidance on manufacturing changes and comparability for human CGT products provides broader recommendations for assessing manufacturing changes in investigational and licensed settings [5].
The critical phrase is low risk. A change is not low risk because it is operationally convenient. It is low risk because the sponsor can explain why the change is unlikely to affect quality, and because the available methods are suitable for detecting the changes that would matter. That judgment is both analytical and manufacturing-based.
Comparability also tests whether the control strategy is biologically grounded. An excellent method for identity may be weak for function. A purity method may miss an altered subpopulation. A potency method may be too variable to detect a small but meaningful shift. Limited comparability data can be persuasive only when the selected assays are aligned with the specific risk introduced by the change.
Process validation flexibility still depends on process understanding
FDA acknowledges that sponsors of CGT products may experience limitations in process performance qualification (PPQ) production. The guidance states that the FDA does not specify a minimum number of PPQ batches in the biologics licensing requirements or current good manufacturing practice (CGMP) regulations, and recommends that sponsors justify the number of PPQ batches based on context-specific factors such as product and process understanding, manufacturing complexity, and the controls in place [1]. The 2011 FDA process validation guidance also frames process validation as a lifecycle activity built on process design, qualification, and continued process verification [7].
This is not a numerical shortcut. If a sponsor cannot produce many PPQ lots, the scientific expectation shifts toward explaining why the available PPQ strategy is sufficient. That explanation should integrate process characterization, in-process controls, raw material controls, release testing, deviations, prior manufacturing history, and continued verification.
Analytical methods are essential here, but they do not stand alone. They must be embedded in a process understanding that explains why measured attributes are stable, controlled, and meaningful. The strongest PPQ rationale is therefore not a statement that three batches are impossible. It is a statement that the process is understood, controlled, and monitored well enough to support the proposed qualification approach. The difference matters because flexibility without process understanding leaves the control strategy vulnerable to unrecognized process or product variability.
Specifications, stability, and rapid methods are lifecycle decisions
Commercial specifications are another area where the guidance acknowledges the realities of CGT development. FDA recognizes that only a small number of CGT product lots may be available at the time of BLA submission, often due to small patient populations. When appropriate, and while still ensuring product quality, the FDA may consider flexible approaches for establishing release specifications when statistically robust commercial acceptance criteria are not feasible at initial approval [1].
FDA also notes that post-approval reevaluation of product release acceptance criteria may be appropriate in certain circumstances as additional commercial batches are produced, while changes to specifications after approval must be submitted in a prior approval supplement [1]. ICH Q6B remains relevant because specifications should reflect tests, procedures, and acceptance criteria suitable for ensuring the quality of biotechnological and biological products [8].
The lesson is not that weak specifications are acceptable. The lesson is that initial specifications for some CGT products may need to be scientifically supported, conservative where appropriate, and explicitly connected to a plan for refinement. A small dataset should not be forced into false statistical certainty. It should be interpreted with transparency about method variability, process history, clinical manufacturing experience, and planned lifecycle learning.
Stability follows the same reasoning. FDA generally recommends real-time stability data from three lots manufactured at the commercial facility with a minimum of six months of data, but the May 2026 guidance says FDA may consider an alternative number of stability lots based on risk evaluation, may consider representative clinical lots, and may consider stability data from similar products as supporting data when a concurrent testing strategy is included [1]. The June 2025 draft guidance from the FDA and ICH Q1 provides the broader stability context for drug substances and drug products [9].
For CGT products, stability is not only an expiration-dating exercise. It is a method suitability exercise. The analytical strategy must detect the changes that matter for the specific product: loss of viability, altered phenotype, loss of vector activity, aggregation, degradation, loss of expression, changes in impurity profile, or other product-specific risks.
Reserve samples and microbiological methods also belong in this lifecycle discussion. FDA recognizes that retaining sufficient material from each biologic lot for six months after expiration may not be feasible for very small or patient-specific CGT lots and may consider exceptions consistent with Title 21 of the Code of Federal Regulations (CFR) section 600.13 [1]. FDA may also consider alternative analytical methods instead of compendial methods when sufficient information and data demonstrate appropriate accuracy, sensitivity, specificity, reproducibility, and suitability for the intended purpose [1]. The 2015 FDA analytical procedures and methods validation guidance provides broader expectations for establishing that analytical procedures are suitable for their intended use [10].
Platforms and standards reduce friction, not responsibility
The May 2026 guidance also recognizes prior knowledge. FDA may consider proposals to leverage CMC knowledge across similar CGT products and critical components, including analytical methods, method validation, lot-release specifications, stability data, comparability data, process development, and process validation [1]. FDA also discusses platform analytical procedures, drawing on the ICH Q2(R2) concept of a method suitable for testing the quality attributes of different products without significant changes to operational conditions, system suitability, or reporting structure [1], [4].
This is one of the most constructive parts of the guidance. Platform methods can reduce avoidable repetition, accelerate development, improve consistency, and make regulatory interactions more predictable. But prior knowledge is not a synonym for assumption. A shared vector class, cell type, construct, unit operation, or analytical technology does not automatically prove that method performance transfers.
The relevant question is narrower and more scientific: is the new product sufficiently similar with respect to the measured attribute and the decision being made?
The same principle applies to voluntary consensus standards. FDA's Standards Recognition Program for Regenerative Medicine Therapies identifies scientifically sound standards that do not conflict with FDA regulations or policy, and the May 2026 guidance notes that using standards can increase regulatory predictability and reduce the amount of documentation needed in a submission [1], [11]. Standards can help align expectations. They cannot replace product-specific understanding when product-specific risk is material.
A more mature CGT CMC ecosystem will likely depend on better platforms: analytical procedures, comparability approaches, reference materials, process understanding, and lifecycle strategies. Their credibility will depend on boundaries. The strongest sponsors will know not only when a platform applies, but when it stops applying.
What a stronger CMC narrative should say
The best response to the FDA's guidance is not a longer CMC checklist. It is a more disciplined scientific argument. In practice, that argument should do four things.
Name the constraint. A limited PPQ strategy, sparse-lot specification, single-lot method validation, rapid microbiological method, or platform analytical procedure should begin with the real development constraint, not a generic appeal to CGT complexity.
Name the risk. The narrative should state which quality concern the conventional evidence package would normally reduce: identity error, impurity risk, potency uncertainty, comparability ambiguity, instability, process drift, or another product-specific risk.
Match evidence to the risk. The sponsor should explain why the selected analytical methods, process controls, prior knowledge, in-process measurements, release data, and stability data are suitable for that risk.
Close the uncertainty. The narrative should define what will be reduced, monitored, or verified later through continued process verification, post-approval specification reevaluation, ongoing stability, post-approval monitoring of commercial batch data, or product-specific verification of platform assumptions.
A strong narrative separates measurement from meaning. It does not assume that more methods automatically create more assurance. It explains why each method exists, which decision the method supports, which failure mode the method can detect, and where the method's limits are. In other words, the goal is not simply more results. The goal is an integrated control strategy.
The sponsor should also separate the FDA's stated flexibility from the sponsor's interpretation. FDA may consider flexible approaches in certain circumstances. The sponsor must explain why those circumstances apply. FDA may consider limited comparability data for certain low-risk changes. The sponsor must explain why the change is low risk and why the data are sufficient. FDA may consider single-lot analytical method validation in appropriate cases. The sponsor must explain why the lot is representative and why the method performance evidence is adequate.
This is where analytical science moves from testing support to regulatory reasoning. It does not merely generate results for a submission. It shapes the reasoning by which evidence generated under constrained development conditions becomes credible.
The real message of the guidance
FDA's May 2026 guidance rejects a false choice. It does not force CGT products into conventional evidence templates when those templates are poorly matched to patient-specific, low-volume, short-shelf-life, or programs with rapidly evolving manufacturing processes. It also does not excuse weak CMC development, even if the product is difficult. It asks for flexible approaches supported by product understanding, process understanding, analytical suitability, risk management, and lifecycle control [1].
That is a demanding standard. It requires sponsors to make sharper scientific arguments when conventional evidence packages are limited or poorly matched to the product. For CGT products, manufacturing innovation will not be enough. The field needs analytical strategies that measure the right attributes, at the right time, with enough confidence to support decisions made under real development constraints. CMC flexibility is not a relaxation of quality. It is a transfer of responsibility onto the quality of the scientific argument. In CGT, that argument will often stand or fall on analytical science, process understanding, and the discipline to know the limits of both.
References
[1] U.S. Food and Drug Administration, Chemistry, Manufacturing, and Controls Flexibilities for Developing Human Cellular and Gene Therapy Products for a Biologics License Application: Guidance for Industry. Silver Spring, MD, USA: FDA, May 2026. Available: https://www.fda.gov/vaccines-blood-biologics/guidance-compliance-regulatory-information-biologics/biologics-guidances
[2] U.S. Food and Drug Administration, Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications: Guidance for Industry. Silver Spring, MD, USA: FDA, Jan. 2020. Available: https://www.fda.gov/media/113760/download
[3] U.S. Food and Drug Administration and International Council for Harmonisation, Q14 Analytical Procedure Development: Guidance for Industry. Silver Spring, MD, USA: FDA, Mar. 2024. Available: https://www.fda.gov/media/161202/download
[4] U.S. Food and Drug Administration and International Council for Harmonisation, Q2(R2) Validation of Analytical Procedures: Guidance for Industry. Silver Spring, MD, USA: FDA, Mar. 2024. Available: https://www.fda.gov/media/161201/download
[5] U.S. Food and Drug Administration, Manufacturing Changes and Comparability for Human Cellular and Gene Therapy Products: Draft Guidance for Industry. Silver Spring, MD, USA: FDA, Jul. 2023. Available: https://www.fda.gov/media/170198/download
[6] U.S. Food and Drug Administration, Potency Assurance for Cellular and Gene Therapy Products: Draft Guidance for Industry. Silver Spring, MD, USA: FDA, Dec. 2023. Available: https://www.fda.gov/media/175132/download
[7] U.S. Food and Drug Administration, Process Validation: General Principles and Practices: Guidance for Industry. Silver Spring, MD, USA: FDA, Jan. 2011. Available: https://www.fda.gov/media/71021/download
[8] U.S. Food and Drug Administration and International Council for Harmonisation, Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. Silver Spring, MD, USA: FDA, Aug. 1999. Available: https://www.fda.gov/media/71510/download
[9] U.S. Food and Drug Administration and International Council for Harmonisation, Q1 Stability Testing of Drug Substances and Drug Products: Draft Guidance for Industry. Silver Spring, MD, USA: FDA, Jun. 2025. Available: https://www.fda.gov/media/187161/download
[10] U.S. Food and Drug Administration, Analytical Procedures and Methods Validation for Drugs and Biologics: Guidance for Industry. Silver Spring, MD, USA: FDA, Jul. 2015. Available: https://www.fda.gov/files/drugs/published/Analytical-Procedures-and-Methods-Validation-for-Drugs-and-Biologics.pdf
[11] U.S. Food and Drug Administration, Voluntary Consensus Standards Recognition Program for Regenerative Medicine Therapies: Guidance for Industry. Silver Spring, MD, USA: FDA, Oct. 2023. Available: https://www.fda.gov/media/159237/download
Additional details
Description
How sponsors can build credible CMC arguments when patient populations are small, manufacturing runs are few, material is limited, and product complexity is high
Identifiers
- GUID
- 198076193
- URL
- https://abrahamfinny.substack.com/p/fdas-2026-cgt-cmc-flexibility-guidance
Dates
- Issued
-
2026-05-17T13:02:46
- Updated
-
2026-05-17T13:02:46
References
- Unknown title https://www.fda.gov/vaccines-blood-biologics/guidance-compliance-regulatory-information-biologics/biologics-guidances
- Unknown title https://www.fda.gov/media/113760/download
- Unknown title https://www.fda.gov/media/161202/download
- Unknown title https://www.fda.gov/media/161201/download
- Unknown title https://www.fda.gov/media/170198/download
- Unknown title https://www.fda.gov/media/175132/download
- Unknown title https://www.fda.gov/media/71021/download
- Unknown title https://www.fda.gov/media/71510/download
- Unknown title https://www.fda.gov/media/187161/download
- Unknown title https://www.fda.gov/files/drugs/published/analytical-procedures-and-methods-validation-for-drugs-and-biologics.pdf
- Unknown title https://www.fda.gov/media/159237/download